







Research
The molecular mechanism of phase separation in disease
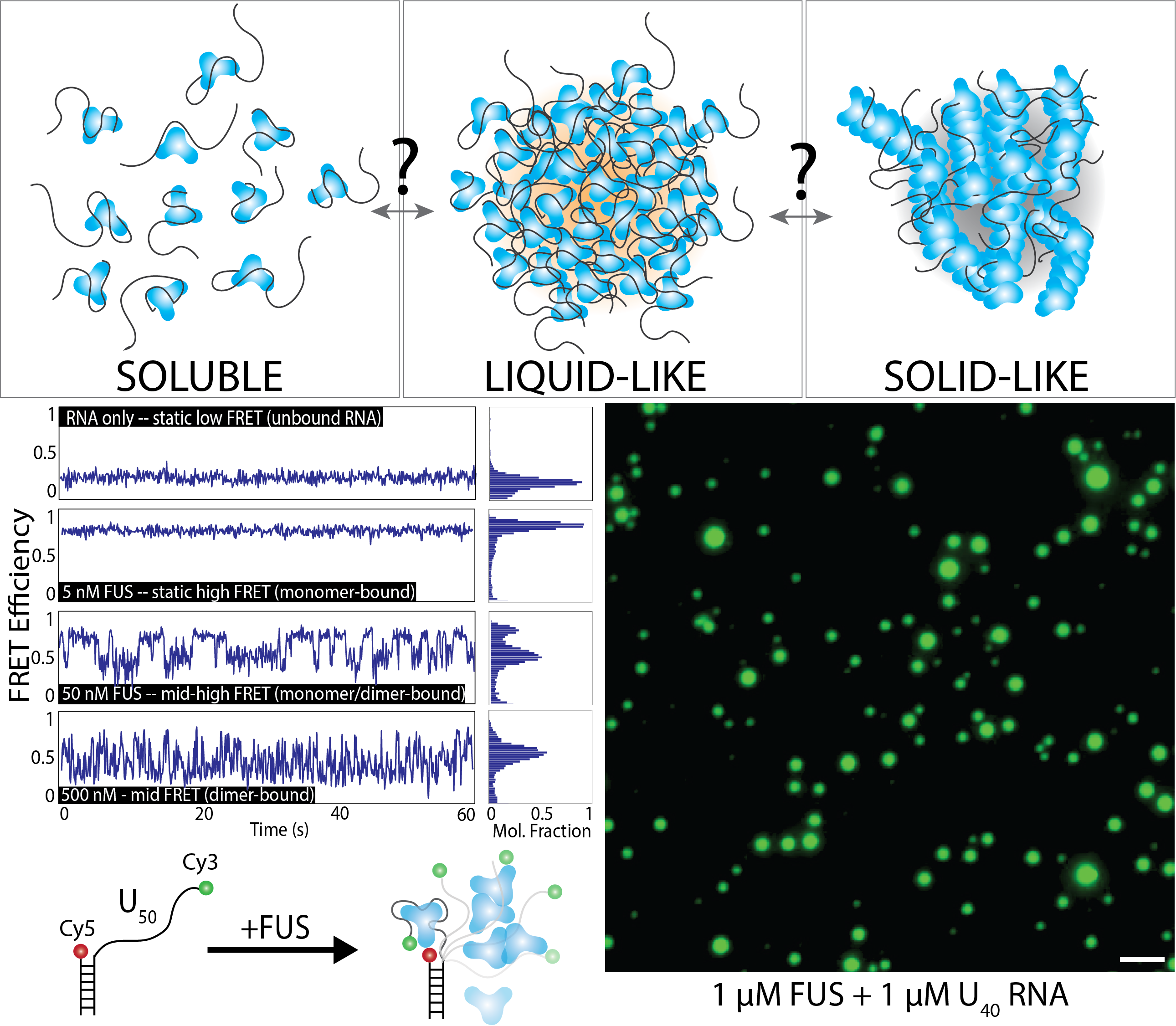
Mutations in the protein Fused in Sarcoma (FUS) have been implicated in ALS (acute lateral sclerosis, also known as Lou Gehrig’s Disease) and FTD (frontotemporal lobal dementia). FUS is an RNA-binding protein with roles in RNA trafficking, splicing, and DNA damage response. Importantly, FUS is known to undergo liquid-liquid phase-separation, which allows the protein to condense into localized droplets that are in a distinct phase from the surrounding environment. In diseased cells, these droplets can solidify into pathogenic aggregates that drive the progression of ALS/FTD. The molecular events that underly the transition from soluble protein to liquid-like RNA-containing droplets to solid-like aggregates remains unknown – likewise, we do not understand how to reverse these aggregation events. Our research is focused on determining the fundamental events that drive nucleation of FUS droplets, especially in the presence of RNA, which is known to drive phase-separation. We use single-molecule techniques such as FRET, PIFE, SMT, optical tweezers, and SimPull to track the oligomerization of phase-separated condensates. We also use bulk techniques such as EMSA, fluorescence anisotropy, and droplet formation to monitor phase-separation. In addition to FUS, we also are interested in TDP-43, Tau, and other proteins linked with neurodegeneration and phase-separation in the context of RNA-binding.
DNA damage and telomere length regulation
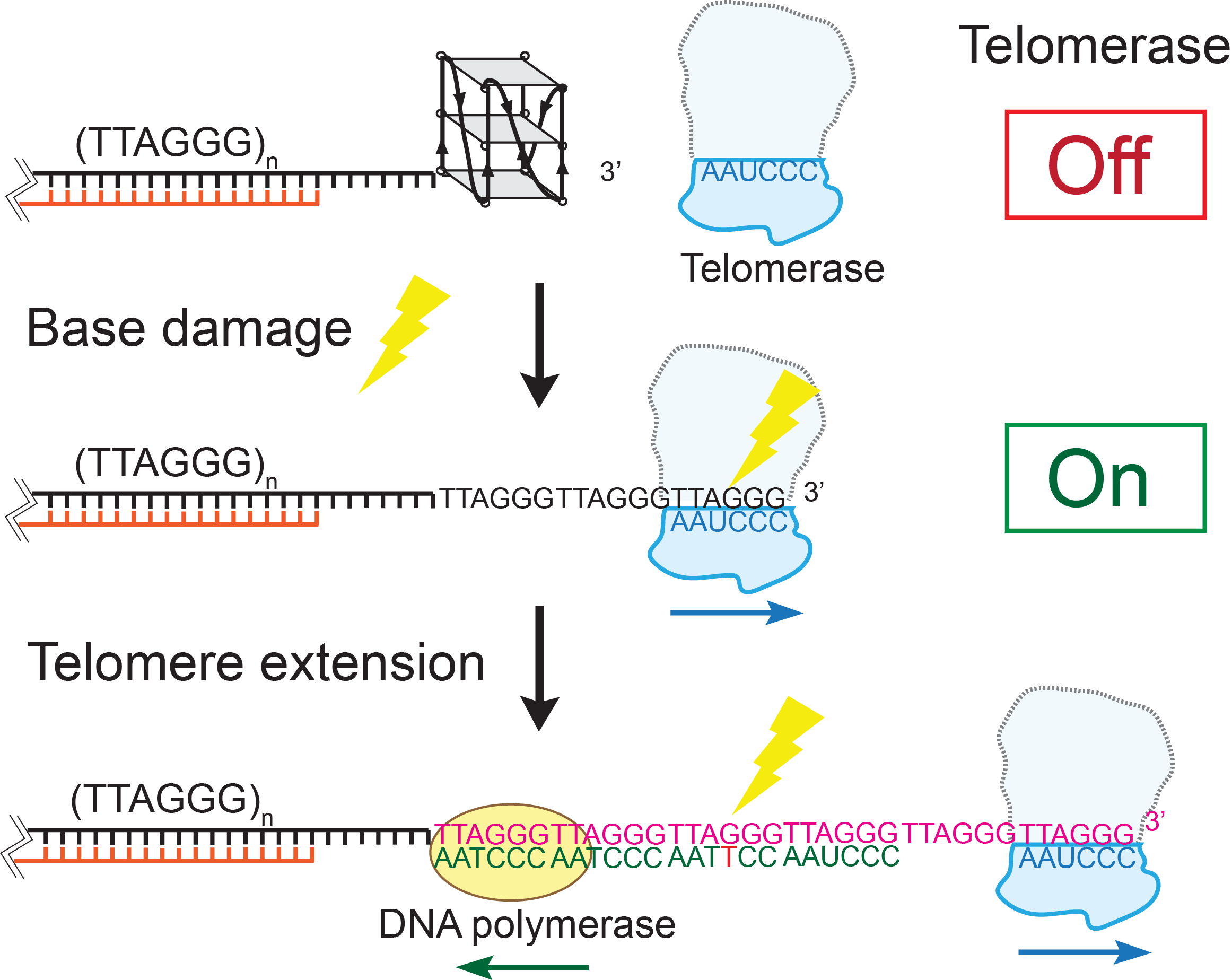
Telomeres are protein-DNA complexes that cap the ends of linear chromosomes. In human cells, telomere DNA backbones are composed of 4-15 kilobase-pairs of repeating 5’-TTAGGG/CCCTAA double-stranded regions and approximately 100 to 200 nucleotides of a single-stranded 5’TTAGGG overhangs at the 3’ end. Specific proteins (collectively called shelterin proteins) bind telomeric DNA to protect it from DNA damage signaling and repair mechanisms, and regulates the length of the telomere. Stem cells and most cancer cells maintain telomere length by activating telomerase, a reverse transcriptase that extends the telomeric overhang by adding TTAGGG tandem repeats. Telomeres are considered hot spots for DNA damage due to their high guanine content. In collaboration with the Opresko laboratory (University of Pittsburgh), we seek to use single-molecule techniques and biochemical assays to investigate how common base modification DNA lesions (such as 8-oxoguanine, thymine glycol, and O6-methylguanine) alter the structure of telomeric single-stranded overhangs, thus modulating shelterin protein binding and telomerase activity. We have showed that the G-quadruplex (GQ) formed in the telomeric overhang blocks telomerase binding. GQ destabilization by base modification lesions leads to increased telomerase binding. Point mutations caused by lesion repair failure can induce the same effect. Our findings suggest a long-term and inheritable effect of telomere DNA damage on telomere lengthening, which potentially contributes to oncogenesis. We are currently studying the effect of telomere base modification and mutation on telomere length regulation.
The role of G-quadruplex forming sequences in the central dogma
G-quadruplex (G4) is a non-canonical secondary structure of DNA or RNA which has been reported in regulating biological processes, including replication, transcription, and translation. It is believed that formation of stable G4 structure can enhance or suppress biological processes, yet the underlying molecular mechanism remains uncertain. We have applied the single molecule FRET method to systematically study G4 forming propensities and the effects on DNA-protein interactions. Currently, we use both in vitro and in vivo assays, coupling with single molecule FRET to elucidate the effect of potential G4 forming sequence (PQS). First, a DNA extension assay is used to quantify the barrier effect of G4 structure, indicating how G4 blocks DNA replication. Second, we applied in vitro transcription assays to examine whether the regulation of transcription is PQS sequence- and/or orientation-dependent. Third, we used both in vitro translation and in vivo E. coli assay to test PQS effect on gene expression. Finally, we devised series of single molecule platforms to dissect stages of transcription including the T7 RNA polymerase binding at promoter, initiation, elongation, progression through PQS and mRNA production. 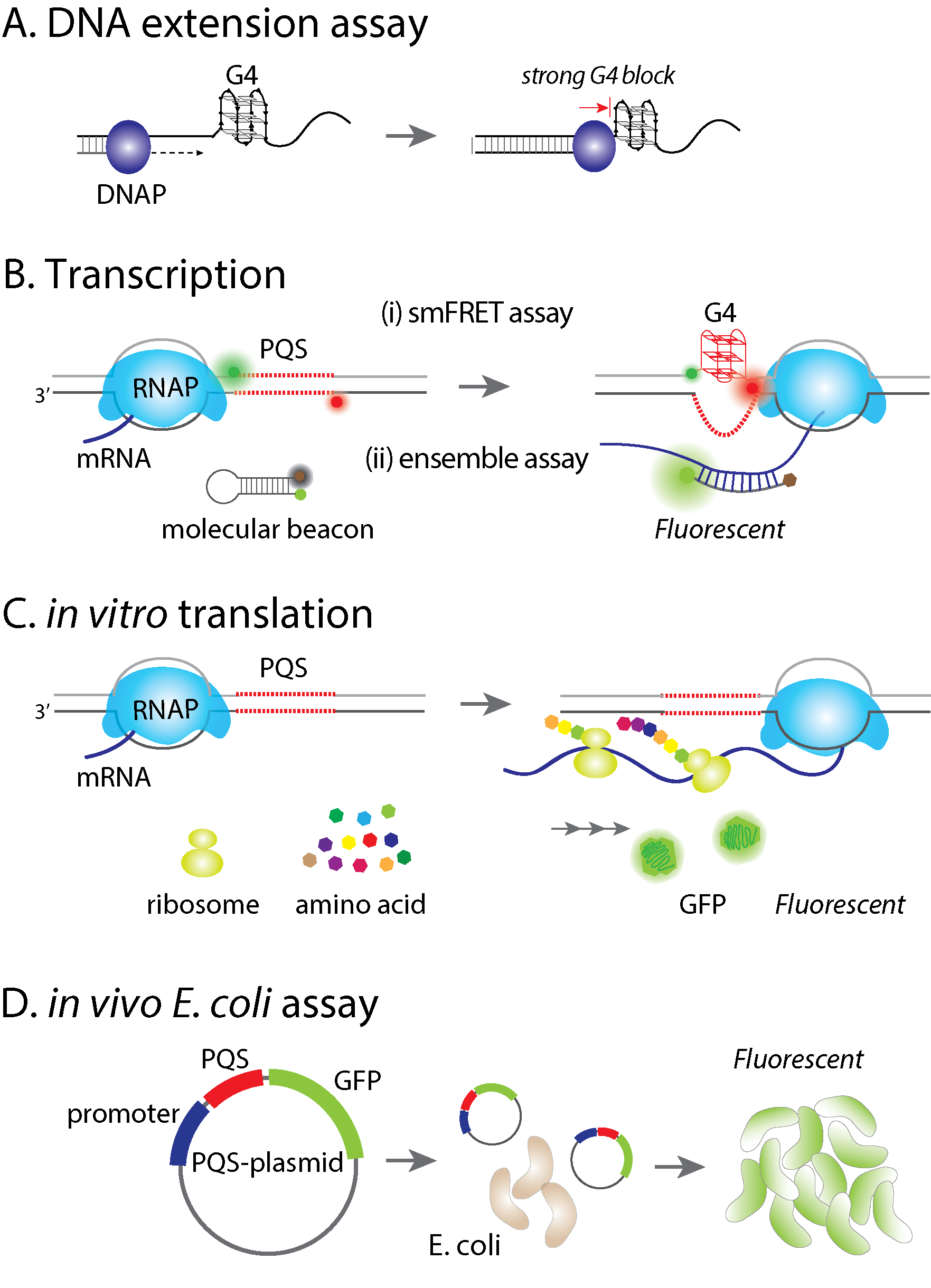
The molecular mechanism of phase separation in diseaseMutations in the protein Fused in Sarcoma (FUS) have been implicated in ALS (acute lateral sclerosis, also known as Lou Gehrig’s Disease) and FTD (frontotemporal lobal dementia). FUS is an RNA-binding protein with roles in RNA trafficking, splicing, and DNA damage response. Importantly, FUS is known to undergo liquid-liquid phase-separation, which allows the protein to condense into localized droplets that are in a distinct phase from the surrounding environment. In diseased cells, these droplets can solidify into pathogenic aggregates that drive the progression of ALS/FTD. The molecular events that underly the transition from soluble protein to liquid-like RNA-containing droplets to solid-like aggregates remains unknown – likewise, we do not understand how to reverse these aggregation events. Our research is focused on determining the fundamental events that drive nucleation of FUS droplets, especially in the presence of RNA, which is known to drive phase-separation. We use single-molecule techniques such as FRET, PIFE, SMT, optical tweezers, and SimPull to track the oligomerization of phase-separated condensates. We also use bulk techniques such as EMSA, fluorescence anisotropy, and droplet formation to monitor phase-separation. In addition to FUS, we also are interested in TDP-43, Tau, and other proteins linked with neurodegeneration and phase-separation in the context of RNA-binding. |
|
|
DNA damage and telomere length regulationTelomeres are protein-DNA complexes that cap the ends of linear chromosomes. In human cells, telomere DNA backbones are composed of 4-15 kilobase-pairs of repeating 5’-TTAGGG/CCCTAA double-stranded regions and approximately 100 to 200 nucleotides of a single-stranded 5’TTAGGG overhangs at the 3’ end. Specific proteins (collectively called shelterin proteins) bind telomeric DNA to protect it from DNA damage signaling and repair mechanisms, and regulates the length of the telomere. Stem cells and most cancer cells maintain telomere length by activating telomerase, a reverse transcriptase that extends the telomeric overhang by adding TTAGGG tandem repeats. Telomeres are considered hot spots for DNA damage due to their high guanine content. In collaboration with the Opresko laboratory (University of Pittsburgh), we seek to use single-molecule techniques and biochemical assays to investigate how common base modification DNA lesions (such as 8-oxoguanine, thymine glycol, and O6-methylguanine) alter the structure of telomeric single-stranded overhangs, thus modulating shelterin protein binding and telomerase activity. We have showed that the G-quadruplex (GQ) formed in the telomeric overhang blocks telomerase binding. GQ destabilization by base modification lesions leads to increased telomerase binding. Point mutations caused by lesion repair failure can induce the same effect. Our findings suggest a long-term and inheritable effect of telomere DNA damage on telomere lengthening, which potentially contributes to oncogenesis. We are currently studying the effect of telomere base modification and mutation on telomere length regulation. |
The role of G-quadruplex forming sequences in the central dogmaG-quadruplex (G4) is a non-canonical secondary structure of DNA or RNA which has been reported in regulating biological processes, including replication, transcription, and translation. It is believed that formation of stable G4 structure can enhance or suppress biological processes, yet the underlying molecular mechanism remains uncertain. We have applied the single molecule FRET method to systematically study G4 forming propensities and the effects on DNA-protein interactions. Currently, we use both in vitro and in vivo assays, coupling with single molecule FRET to elucidate the effect of potential G4 forming sequence (PQS). First, a DNA extension assay is used to quantify the barrier effect of G4 structure, indicating how G4 blocks DNA replication. Second, we applied in vitro transcription assays to examine whether the regulation of transcription is PQS sequence- and/or orientation-dependent. Third, we used both in vitro translation and in vivo E. coli assay to test PQS effect on gene expression. Finally, we devised series of single molecule platforms to dissect stages of transcription including the T7 RNA polymerase binding at promoter, initiation, elongation, progression through PQS and mRNA production. | |